
Introduction
Many gram-negative bacteria infect hosts and cause diseases by translocating a variety of type III secreted effectors (T3SEs) into host cytoplasm. However, it remains challenging for accurate prediction of T3SEs. Traditional computational models mainly depend on the sequence-based atypical features buried in the N-terminal peptides of T3SEs, but tend to have a high false positive rate.
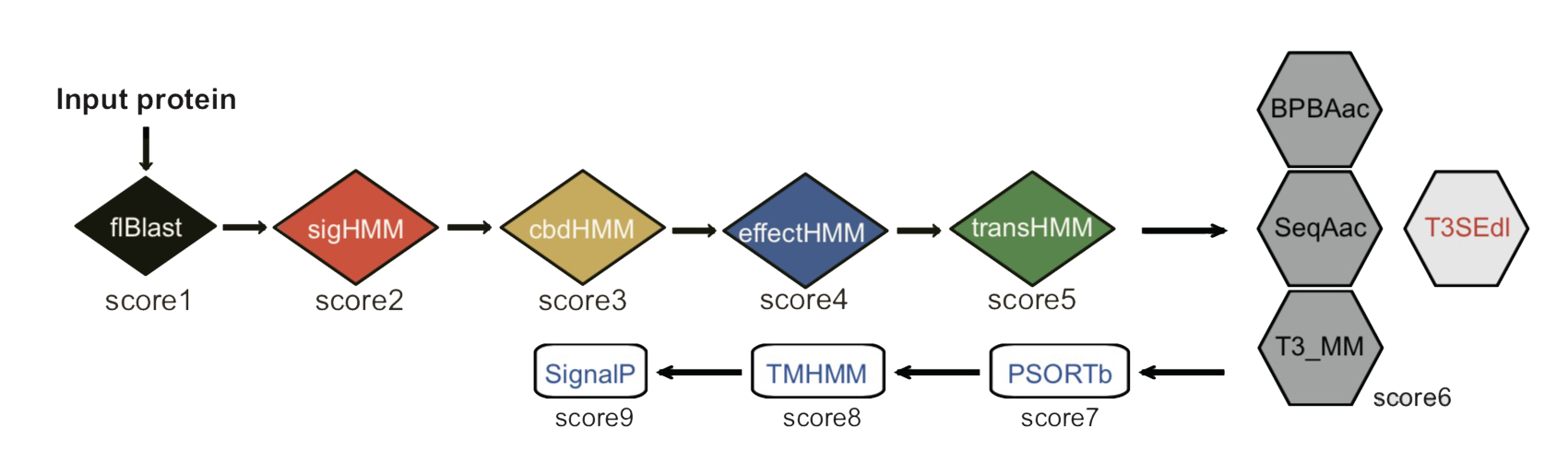
To increase the precision of prediction, we made a comprehensive survey on multiple aspects of the biological property of T3SEs, including the signal regions, chaperone-binding domains, effector domains and gene promoters, and integrated these features for T3SE prediction. Furthermore, a list of machine-learning algorithms, including Markov Model, Support Vector Machine, Decision Tree and Deep Learning, were adopted to train the sequential or position-specific frequency, conditional probability, mono-/bi-/tri-profile Aac represented atypical features buried in the signal regions of T3SEs. A voting-based ensemble model integrated the prediction results of individual machine-learning models. Finally, we assembled a unified pipeline, T3SEpp, which used a linear model to integrate the results of modules detecting the comprehensive features, achieved good performance (accuracy of ~0.94), and remarkably reduced the false positive rate by ~10 folds without apparent loss of sensitivity. The sequence features observed here and the T3SEpp pipeline would facilitate accurate identification of new T3SEs and the bacteria-host interaction studies.
A webserver was launched to implement the T3SEpp automatically. Alternatively, users could also download the standalone version of T3SEpp to make the prediction in a personal computer.